本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如若转载,请注明出处:http://www.ldbm.cn/p/440233.html
如若内容造成侵权/违法违规/事实不符,请联系编程新知网进行投诉反馈email:809451989@qq.com,一经查实,立即删除!相关文章

MySQL record 01 part
更改密码: alter user rootlocalhost identified with mysql_native_password by ‘123456’; 注意:
在命令行方式下,每条MySQL的命令都是以分号结尾的,如果不加分号,MySQL会继续等待用户输入命令,直到MyS…

【最新华为OD机试E卷-支持在线评测】跳马(200分)多语言题解-(Python/C/JavaScript/Java/Cpp)
🍭 大家好这里是春秋招笔试突围 ,一枚热爱算法的程序员 ✨ 本系列打算持续跟新华为OD-E/D卷的三语言AC题解 💻 ACM金牌🏅️团队| 多次AK大厂笔试 | 编程一对一辅导 👏 感谢大家的订阅➕ 和 喜欢💗 🍿 最新华为OD机试E卷,全、新、准,题目覆盖率达 95% 以上,支持…

图片无损缩放PhotoZoom Pro 9.0.2绿色版 +免费赠送PhotoZoom激活优惠代码
PhotoZoom Pro 9.0.2 是一款专业的图片无损缩放软件,该软件采用了 benvista s-spline 独特技术,增强了对图像格式的支持,多处理器支持,GPU 加速,win10和 Photoshop CC 支持。带来一流的数字图形扩展与缩减技术。该软件…

6.1.数据结构-c/c++堆详解下篇(堆排序,TopK问题)
上篇:6.1.数据结构-c/c模拟实现堆上篇(向下,上调整算法,建堆,增删数据)-CSDN博客
本章重点
1.使用堆来完成堆排序
2.使用堆解决TopK问题
目录
一.堆排序
1.1 思路
1.2 代码
1.3 简单测试 二.TopK问…
微信小程序原生支持TS、LESS、SASS能力探究
文章目录 原生支持开始使用旧项目新建项目TS声明文件更新 功能说明less 使用全局变量sass 使用全局变量 可以参考原文
在之前开发小程序中,无法使用 less/sass 等 css 预编译语言,也无法使用 TS 进行开发,但在最新的编辑器版本中,…
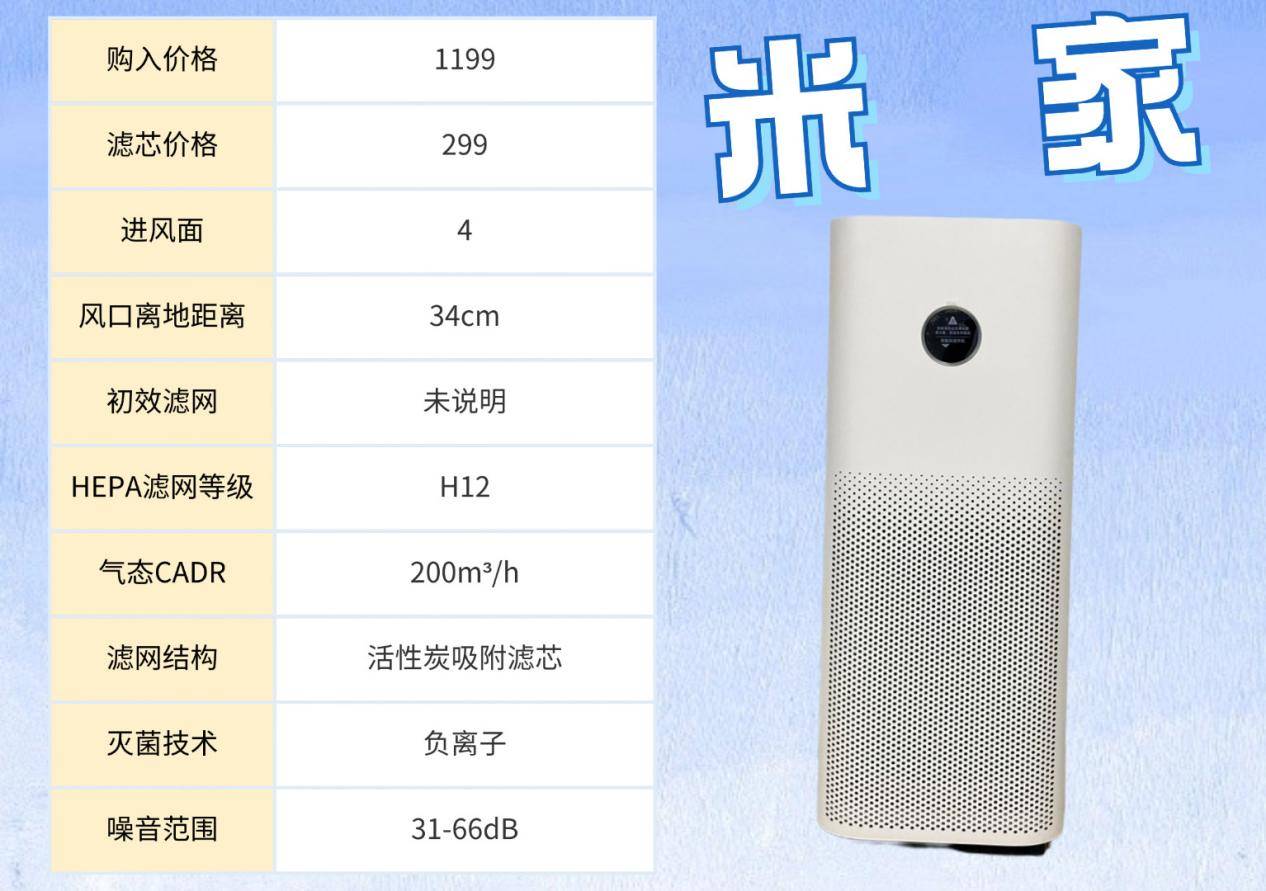
怎么样处理浮毛快捷又高效?霍尼韦尔、希喂、米家宠物空气净化器实测对比
掉毛多?掉毛快?猫毛满天飞对身体有危害吗?多猫家庭经验分享篇: 一个很有趣的现象,很多人在养猫、养狗后耐心都变得更好了。养狗每天得遛,养猫出门前得除毛,日复一日的重复磨练了极好的耐心。我家…

AI驱动的Web3革命:如何通过智能技术实现去中心化
在数字化进程飞速发展的今天,Web3正成为互联网的未来,通过去中心化理念重塑我们的数字世界。与此同时,人工智能(AI)的引入进一步推动了Web3的发展,使其在效率、安全性和用户体验上得到了显著提升。本文将详…

什么是勒索病毒?如何防止勒索病毒入侵
什么是勒索病毒?
最近几年,勒索病毒“异军突起”让本就严峻的数据安全更是雪上加上霜,几乎是每隔几天,就会有企事业单位中招。勒索病毒,是一种性质恶劣、危害极大的电脑病毒,主要通过邮件、木马、网页挂马…


构建数字产业生态链,共绘数字经济新蓝图
在当今数字化浪潮席卷全球的时代,构建数字产业生态链成为了推动经济发展的关键引擎。数字产业生态链如同一个强大的磁场,吸引着各类创新要素汇聚,共同描绘出数字经济的宏伟新蓝图。 数字产业生态链的核心在于融合与协同。它将软件开发、数据分…

安全运营之浅谈SIEM告警疲劳
闲谈: 刚开始学习SIEM、态势感知这类产品的时,翻阅老外们的文章总是谈什么真阳性,假阳性告警、告警疲劳,当时在国内资料中没找到很合理的解释,慢慢就淡忘这件事了。随着慢慢深入工作,感觉大概理解了这些概念…

加密货币市场持有与价格波动:CFI调查揭示的趋势与未来展望
自2022年1月以来,消费者金融协会(CFI)通过六项不同的调查收集了有关加密货币所有权的数据。这些调查旨在了解加密货币的当前持有量和未来购买兴趣,并将这些数据与加密货币市场表现进行对比。结果显示,市场价格与持有量…

【Python】Python 读取Excel、DataFrame对比并选出差异数据,重新写入Excel
背景:我在2个系统下载出了两个Excel,现在通过对下载的2个Excel数据,并选出差异数据 从新写入一个新的Excel中 differences_url rC:\Users\LENOVO\Downloads\differences.xlsx; //要生成的差异Excel的位置及名称
df1_url rC:\Users\LENOVO\Dow…

110001安庆巡检_工艺巡检
安庆巡检_工艺巡检 一. 工艺配置二. 点检计划三. 点检任务四. 复检任务1. 复检列表1.1 页面展示 2. 复检任务下发2.1 操作说明2.2 业务说明2.3 表关联说明ps_recheck_task工艺工序参数_复检详情表 3. 复检详情2.1 获取参数点检详情2.2 获取复检详情列表 4. app端复检任务提交4.…

一文解答Swin Transformer + 代码【详解】
文章目录 1、Swin Transformer的介绍1.1 Swin Transformer解决图像问题的挑战1.2 Swin Transformer解决图像问题的方法 2、Swin Transformer的具体过程2.1 Patch Partition 和 Linear Embedding2.2 W-MSA、SW-MSA2.3 Swin Transformer代码解析2.3.1 代码解释 2.4 W-MSA和SW-MSA…


算法入门-深度优先搜索3
第六部分:深度优先搜索
112.路径总和(简单)
题目:给你二叉树的根节点 root 和一个表示目标和的整数 targetSum 。判断该树中是否存在 根节点到叶子节点 的路径,这条路径上所有节点值相加等于目标和 targetSum 。如果…

Self Refine技术测评:利用Self Refine提高LLM的生成质量
1. 背景与挑战
在当今人工智能蓬勃发展的时代,大型语言模型(Large Language Models,简称 LLMs)已成为众多企业不可或缺的核心技术。从智能客服到内容创作,LLMs 在各个领域都展现出了惊人的能力。然而,随着…
电机驱动及编码器测速(基于STM32F103C8T6HAL库)
硬件:STM32F103C8T6、电机驱动模块tb6612、25GA370带编码器测速盘直流减速电机。 1.电机驱动
1.1 电机驱动模块tb6612
(1)电机驱动模块tb6612简介
电机驱动需要使用电机驱动模块,电机驱动模块把3.3V的电机信号转换成12V的电机的…

【MySQL00】【 杂七杂八】
文章目录 一、前言二、MySQL 文件1. 参数文件2. 日志文件3. 套接字文件4. pid 文件5. 表结构定义文件6. InnoDB 存储引擎文件 二、BTree 索引排序三、InnoDB 关键特性1. 插入缓冲1.1 Insert Buffer 和 Change Buffer1.1 缓冲合并 2. 两次写2. 自适应哈希索引3. 异步IO4. 刷新邻…