本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如若转载,请注明出处:http://www.ldbm.cn/p/440076.html
如若内容造成侵权/违法违规/事实不符,请联系编程新知网进行投诉反馈email:809451989@qq.com,一经查实,立即删除!相关文章
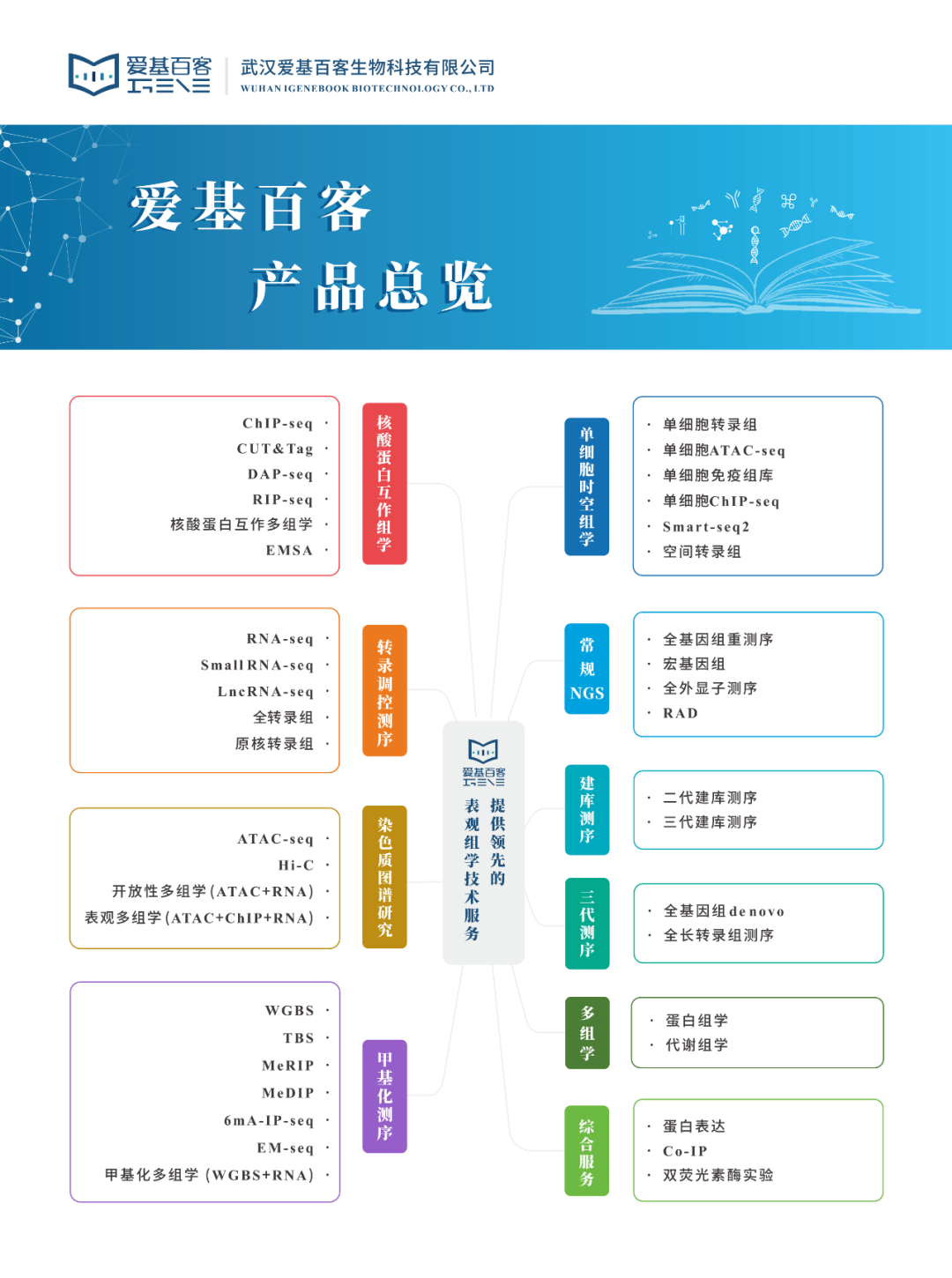
干货!如何利用scRNA数据对空间转录组进行注释
单细胞RNA测序(scRNA-seq)识别组织内的细胞亚群,但不能捕获它们的空间分布,也不能揭示细胞间通讯的局部网络。空间转录组能够对细胞进行定位,却无法准确地在组织中产生深层单细胞分辨率的转录组图谱。成功整合单细胞和空间转录组数据的分析将…

EPIC超级“喜加N”!AOC U32G4ZMN助你畅玩缤纷九月
废土探险、绿茵征战、狙击猎杀,AOC U32G4ZMN带你玩转九月!
Epic游戏商城近日开启“喜加N”游戏盛宴,上周公布的免费游戏:《辐射经典合集》与《外卡橄榄球》,领取时间截止至9月5日,还没领取的玩家ÿ…

六、MySQL高级—架构介绍(1)
🌻🌻 目录 一、Mysql 简介1.1 概述1.2 Mysql 高手是怎样炼成的 二、Mysql Linux 版的安装2.1 mysql5.52.2 mysql5.7 三、Mysql 的用户与权限管理3.1 MySQL的用户管理3.2 权限管理3.3 通过工具远程访问 四、 Mysql的一些杂项配置(了解)五、 Mysql 逻辑架构…

设计模式-面试题(工厂方法模式、策略模式和责任链模式)
开闭原则:扩展开放、修改关闭 工厂设计模式:解耦 简单工厂模式 CoffeeStore和SimpleCoffeeFactory的耦合、SimpleCoffeeFactory和Coffee的耦合 后续如果再加新品种的咖啡,需要修改SimpleCoffeeFactory,这样就违反了开闭原则 简单…

【话题讨论】IBM中国研发部裁员:全球化竞争下的战略调整与行业启示
目录
引言
一、整体分析
1.1 IBM裁员背景与原因
1.2 可能带来的影响
1.3 全球IT产业格局变化趋势
二、人才发展
2.1 对中国IT人才市场的影响
2.2 IT从业者提升自身竞争力的建议
三、产业未来
3.1 应对跨国公司战略调整的策略
3.2 提升自主创新能力的路径
3.3 打造核…

OmniGlue论文详解(特征匹配)
OmniGlue论文详解(特征匹配) 摘要1. 引言2. 相关工作2.1. 广义局部特征匹配2.2. 稀疏可学习匹配2.3. 半稠密可学习匹配2.4. 与其他图像表示匹配 3. OmniGlue3.1. 模型概述3.2. OmniGlue 细节3.2.1. 特征提取3.2.2. 利用DINOv2构建图形。3.2.3. 信息传播与…

虚拟现实智能家居实训系统实训解决方案
随着科技的飞速发展,智能家居已成为现代生活的重要组成部分,它不仅极大地提升了居住的便捷性与舒适度,还推动了物联网、大数据、人工智能等前沿技术的融合应用。为了满足市场对智能家居专业人才日益增长的需求,虚拟现实智能家居实…

《数字信号处理》学习05-单位冲击响应与系统响应
目录
一,单位冲激响应
二,LTI系统对任意序列的系统响应 三,LTI系统的性质 通过上一篇文章《数字信号处理》学习04-离散时间系统中的线性时不变系统-CSDN博客的学习,我已经知道了离散时间线性时不变系统(LTI&#x…

铝材的知识与应用,基础全面
铝的基本性质
银白色,在潮湿的空气中能形成一层防止金属腐蚀的氧化膜,相对密度2.7g/cm3,熔点660℃,沸点2327℃,比强度较高,有良好的导电性和导热性,高反射性和耐氧化性。 二、种类
型材类&…
![[ios]准备好app后使用xcode发布ios操作](https://i-blog.csdnimg.cn/direct/2c5a0c30fe30452eb7c34cf208231946.png)
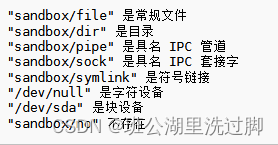
标准库标头 <filesystem> (C++17)学习之文件类型
本篇介绍filesystem文件库的文件类型API。 文件类型 is_block_file (C17) 检查给定的路径是否表示块设备 (函数) is_character_file (C17) 检查给定的路径是否表示字符设备 (函数) is_directory (C17) 检查给定的路径是否表示一个目录 (函数) is_empty (C17) 检查给定的路径是…

VMware Fusion Pro 13 Mac版虚拟机 安装Win11系统教程
Mac分享吧 文章目录 Win11安装完成,软件打开效果一、VMware安装Windows11虚拟机1️⃣:准备镜像2️⃣:创建虚拟机3️⃣:虚拟机设置4️⃣:安装虚拟机5️⃣:解决连不上网问题 安装完成!࿰…

港科夜闻 | 叶玉如校长出席2024科技+新质生产力高峰论坛发表专题演讲,贡献国家科技强国战略...
关注并星标 每周阅读港科夜闻 建立新视野 开启新思维 1、叶玉如校长出席“2024科技新质生产力高峰论坛”,做了题为“三个创新:培育和发展新质生产力、贡献国家科技强国战略”的主题演讲。该论坛于9月2日在香港召开。论坛围绕夯实基础科研、推动源头创新、…
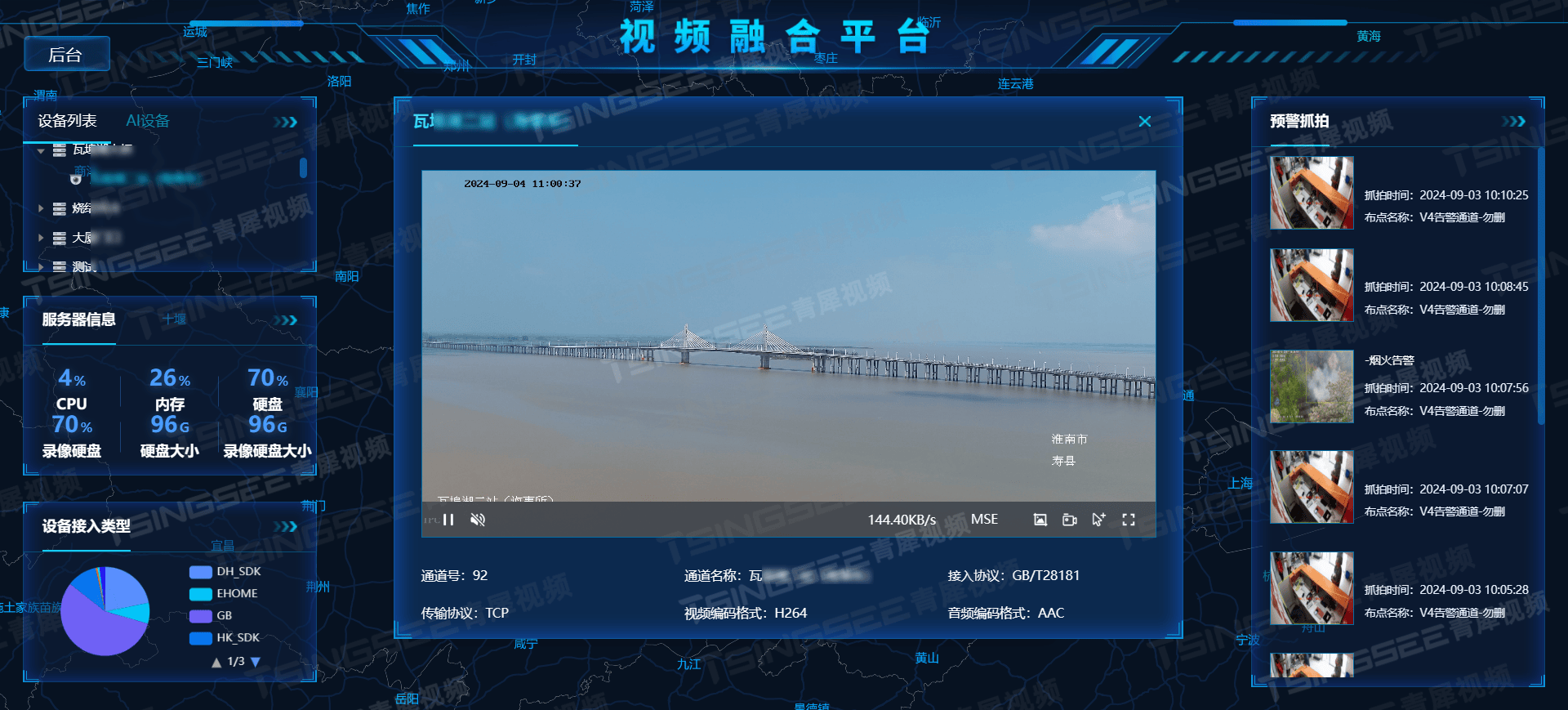
打造安心宠物乐园:EasyCVR平台赋能猫咖/宠物店的智能视频监控解决方案
随着宠物经济的蓬勃发展,宠物店与猫咖等场所对顾客体验、宠物安全及健康管理的需求日益提升。然而,如何确保这些场所的安全与秩序,同时提升顾客体验,成为了经营者们关注的焦点。引入高效、智能的视频监控方案,不仅能够…

微信企业微信忽然爆满 怎么清理才干净?一招彻底清理干净垃圾文件
大家都知道,微信用久了就会堆积很多的垃圾,然后导致系统空间不足,继而引发一些不必要的系统问题,所以清理掉这些垃圾是有必要的。今天我们就给大家介绍一个可以清理微信和企业微信的工具。
解决微信&企业微信忽然爆满的步骤&…

PMP–一、二、三模–分类–14.敏捷–技巧–变革就绪情况
文章目录 技巧一模14.敏捷–组织考虑因素--变革就绪情况--管理层的变更意愿,是组织是否变革就绪的重要前提。答案关键字,获得支持107、 [单选] 一家沉浸于传统瀑布式项目管理中的PMO聘请了你,作为敏捷实践者,来指导组织向敏捷的转…

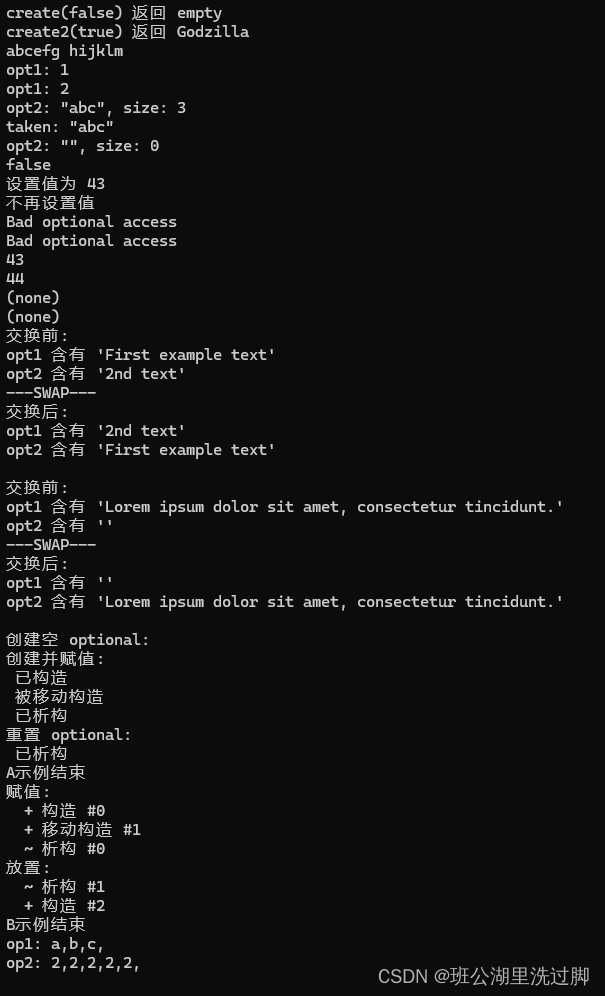
标准库标头 <optional> (C++17)学习之optional
类模板 std::optional 管理一个可选 的所含值,即既可以存在也可以不存在的值。
一种常见的 optional 使用情况是作为可能失败的函数的返回值。与如 std::pair<T, bool> 等其他手段相比,optional 可以很好地处理构造开销高昂的对象&a…

数据库的操作:SQL语言的介绍
一.前言
SQL是一种结构化查询语言。关系型数据库中进行操作的标准语言。
二.特点
①对大小写不敏感
例如:select与Select是一样的
②结尾要使用分号 没有分号认为还没结束;
三.分类
①DDL:数据定义语言(数据库对象的操作(结…

通义千问更新数学大模型及视觉多模态
Qwen2-Math,这是通义千问专门为数学场景优化的模型,其数学能力指标甚至超越了GPT4o, Claude3.5 Sonnet, Deepseek Coder等顶流模型,目前从指标来看是最强的数学模型。目前是免费供应,大家碰到数学问题可以选择使用这个模型。 Qw…